无缝克隆试剂盒多片段
- D0204P-25T
- Lablead/兰博利德
- 北京市
- 一周内
- 25T定制
- 650 元
- 2022-07-29 15:48:51
北京兰博利德商贸有限公司
DNA Assembly Mix Plus
产品货号:D0204P
储存条件:-20℃
产品组分
|
组分/规格 |
10T |
25T |
50T |
|
DNA Assembly Mix PLUS |
50μl |
125μl |
250μl |
|
pUC19 Control Plasmid, Linearized (Ampr, 40 ng/μl) |
5μl |
5μl |
5μl |
|
500 bp Control Fragment (20 ng/μl) |
5μl |
5μl |
5μl |
产品简介
基于重组原理的无缝克隆技术,作为新一代的克隆方法,不依赖繁琐的酶切、连接步骤,也不需要末端补平等操作,依据DNA片段与线性化载体末端的15~25nt同源序列的重组,可将插入片段克隆至任意线性载体的任意位点,载体自连背景极低,是一种简单、快速、高效的DNA定向克隆技术。
DNA Assembly MixPLUS无缝克隆试剂盒最快仅需5分钟即可完成单片段重组,且阳性率高于95%。Mix中添加的辅助因子,有效提高克隆阳性率;经优化的反应体系,能够一定程度上耐受未纯化PCR产物中含有的杂质。升级版的无缝克隆试剂盒具有更高的阳性率、更好的兼容性。
使用简单 —— 兼容PCR与酶切体系,省去繁琐的纯化步骤
超高效率 —— 可以稳定支持5-6片段在同一克隆体系
稳定可靠 —— 37℃放置一周或冻融30次,无明显下降
实验步骤
1、实验流程概要
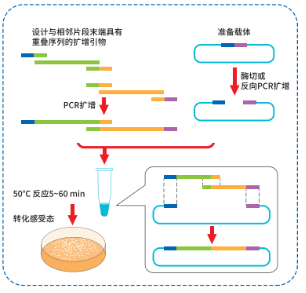
2、线性化克隆载体制备
选择合适的克隆位点,对载体进行线性化,载体的线性化可以通过酶切或反向 PCR 扩增完成。
① 酶切制备
推荐使用LabFD™快速内切酶进行双酶切,使载体线性化完全,以降低转化背景(假阳性克隆);若使用单酶切进行线性化,可以适当延长酶切时间以减少环状质粒残留。
注1:经双酶切进行线性化的载体无需去磷酸化,经单酶切则需要去磷酸化;
注2:酶切完成后,应将快速内切酶失活或对目的产物纯化后再用于重组反应。
② 反向 PCR 扩增制备
为减少扩增突变的引入,推荐使用高保真PCR Mix进行扩增。推荐使用预线性化质粒作为模板,以减少环状质粒模板残留对克隆阳性率的影响。
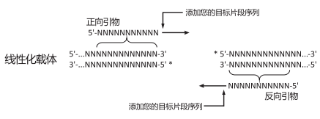
3、插入片段PCR引物设计
PCR引物的5' 端必须包含与其相邻片段(插入片段或载体)末端同源的15~25 nt(推荐18nt)序列。假如载体为粘性末端,且3'端突出,则引物设计必须包含突出部分;若5'端突出,则引物设计可以包含突出部分,也可以不包含。
插入片段正向扩增引物:
5'—上游载体末端同源序列+酶切位点(可选)+ 基因特异性正向扩增序列— 3'
插入片段反向扩增引物:
3'—基因特异性反向扩增序列+酶切位点(可选)+下游载体末端同源序列—5'
注1:尽量选择无重复序列且GC含量均匀的区域进行克隆,当载体克隆位点上下游25 nt区域内GC含量为40%~60%时,重组效率最高;
注2:本试剂盒所提供的 pUC 19 载体(Ampr)连接端序列如下:
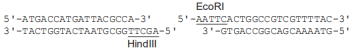
4、插入片段的 PCR 扩增
推荐使用高保真PCR Mix进行扩增,以减少扩增突变的引入。建议使用纯化后的PCR产物进行无缝克隆反应,若PCR产物经琼脂糖凝胶电泳鉴定为特异性扩增产物,可直接使用,但加样体积不应超过总反应体积的20%。
5、重组反应
①于冰水浴中配置以下反应体系:
|
组分 |
反应体系 |
阴性对照 |
阳性对照(如有必要) |
|
DNA Assembly Mix |
5μl |
5μl |
5μl |
|
线性化载体(ng) |
50~200ng |
50~100ng |
pUC19 Control Plasmid,Linearized, 1 μl |
|
插入片段 |
10~200 ng |
- |
500 bp Control, Fragment, 1 μl |
|
ddH2O |
To 10 μl |
||
a.最适载体用量(ng)=0.02×载体碱基对数,即0.03pmol。
b.插入单片段,最适片段用量(ng)= 0.04×片段碱基对数;
插入多片段,每片段最适用量(ng)= 0.02×片段碱基对数。
注1:若插入单片段的长度大于载体,则应互换载体与插入片段用量;
注2:若插入片段的长度小于200 bp,则插入片段应使用5倍载体用量;
注3:若按上述公式计算得到的用量超过最低/最高值,则建议直接按最低/最高用量使用;
注4:载体片段过长、插入片段过长克隆阳性率均会降低。
重组反应体系配制完成后,用移液枪轻轻吸打混匀各组分,避免产生气泡,切勿涡旋。
② 将反应体系置于50℃,反应5~60min。
注1:推荐使用 PCR 仪等温控比较精准的仪器进行反应,反应时间不足或太长克隆效率均会降低;
注2:插入1-2个片段时,推荐反应时间5-15min,插入3-5个片段时,推荐反应时间15-30min;
注3:当载体骨架在 10 kb 以上或插入片段在4 kb以上时,建议延长反应时间到30-60min
注4:50℃反应完成后,建议进行瞬时离心,将反应液收集至管底。
③ 将反应液离心管置于冰上冷却,之后进行转化或者储存于-20℃。
注1:-20℃储存的重组产物,建议在1周内使用。
6、重组产物转化
取5~10μl反应液,加入到100 μl感受态细胞中缓慢吸打混匀,冰上放置30min。42℃热激45~60sec,冰浴5 min。加500 μl SOC或LB培养基,37℃振荡培养40~60min(200 rpm)。将菌液均匀涂布在含有对应抗生素的平板上,倒置于37℃过夜培养。
注1:不同感受态细胞最后的克隆阳性率会有所差别,推荐使用转化效率>108 CFU/μg的感受态细胞;
注2:菌落数取决于PCR产物与线性化载体的数量和纯度;
注3:阳性对照平板通常生长大量白色单菌落,阴性对照平板只生长很少的菌落。
7、阳性克隆检测
挑取单菌落至10 μl ddH2O中混匀,95℃裂解10 min 后,取1 μl裂解液作为模板,进行菌落PCR鉴定,或将单菌落接种至抗性培养基中培养过夜后,提取质粒进行酶切鉴定。
注1:菌落 PCR 时建议至少使用一条通用引物,避免假阳性结果;
注2:必要时可进一步对阳性结果进行测序鉴定。
常见问题
|
问题描述 |
原因 |
解决方法 |
|
转化效率低 |
感受态效率低下 |
使用新制备或妥善保存的感受态细胞。 |
|
DNA片段比例不佳 |
按照说明书推荐的最适用量和比例配制反应体系。载体和插入片段的浓度测定: 若线性化载体与插入片段已经过纯化,且经电泳检测条带单一或无Smear残留时,可使用超微量核酸蛋白检测仪等基于分光光度法的仪器进行浓度测定,但只有当A260/A280 在1.8~2.0之间时浓度值可信;若线性化载体与插入片段未经过纯化,也可使用琼脂糖电泳测定样品浓度。 |
|
|
DNA片段纯度不够 |
对载体和插入片段进行胶回收纯化。由于EDTA等金属离子螯合剂会抑制无缝克隆反应,因此纯化产物应溶解于ddH2O中,切勿使用Tris-EDTA等缓冲液。 |
|
|
反应产物过量 |
在转化体系中,无缝克隆反应产物体积不应超过感受态细胞体积的10%。 |
|
|
大量克隆不含插入片段 |
载体线性化不完全 |
酶切制备线性化载体时,提高快速内切酶的使用量,延长反应时间,使用胶回收纯化酶切产物。 |
|
相同抗性质粒污染 |
以质粒为模板进行插入片段 PCR 扩增时,使用预线性化质粒作为扩增模板,使用DpnI 等甲基化敏感型内切酶对扩增产物进行处理,或对产物进行胶回收纯化。 |
|
|
平板抗性不足 |
确保使用正确的抗生素,并使用新鲜制备的抗生素平板。 |
|
|
大量克隆含有 不正确插入片段 |
非特异性PCR扩增产物 |
优PCR体系,提高扩增特异性,或胶回收纯化PCR产物重叠序列的扩增引物。 |