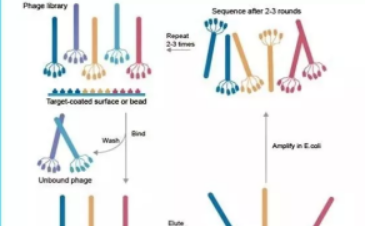
噬菌体文库构建
噬菌体文库
1. 实验目的
使用抗原免疫后羊驼PBMC构建噬菌体文库
2. 实验设计
从羊驼PBMC中提取总RNA,反转录为cDNA,经过两轮PCR扩增后得到抗体序列扩增产物,选择载体进行酶切后连接,最后转化至SS320大肠杆菌感受态细胞得到细菌文库,经过辅助噬箘体(M13KO7)侵染并诱导后制备获得噬菌体文库。
3. 实验方法与步骤
3.1总RNA提取
3.1.1将羊驼PBMC样品从-80℃冰箱中取出,分装0.5ml/管,每管中加入100μl氯仿,在震荡器上剧烈震荡15s后,室温放置静置5min;
3.1.2将离心机预冷至4℃,12000g离心15min,离心后出现分层,将最上层的透明层用移液枪小心转移到新的无RNase和DNase的1.5ml离心管中,每管加入250μl的异丙醇,上下颠倒多次混匀后室温放置静置10min;
3.1.3 12000g离心10min,去除上清保留沉淀;
3.1.4每管加入1ml 75%乙醇,上下颠倒多次以悬浮沉淀;
3.1.57500g离心5min,去除上清保留沉淀;
3.1.6保持离心管口打开,室温放置干燥10min,每管加入DEPC处理水溶解,55℃孵育10min以确保RNA完全溶解;
3.1.7小心吸取至同一个离心管中,即为PBMC中提取出的总RNA;
3.1.8取1μl上述总RNA进行1%琼脂糖凝胶电泳以检测总RNA的完整性,同时取2μl用核酸浓度测量仪检测RNA的浓度和A260/A280并记录。
3.2 RNA反转录
样品在总RNA提取后立即使用反转录试剂盒进行反转录以减少RNA的降解。
3.2.1将上述总RNA样品分为两份,一份使用试剂盒内的Oligo dT Primer作为引物,另一份用试剂盒内的Random 6-mers作为引物,在1.5ml离心管中按下表比例配置反应液,当总RNA量大于5μg时,同比例扩大反应体系。
|
试剂 |
使用量 |
|
Oligo dT Primer(50μM)或者 Random 6-mers(50μM) |
1μl |
|
dNTP Mix |
1μl |
|
总RNA |
5μg |
|
ddH2O |
Up to 10μl |
3.2.2将含有上述反应液的离心管放入金属加热器中,65℃反应5min使RNA变性,反应结束后置于冰上迅速冷却;
3.2.3在上述1.5ml离心管中按下表比例配置反应液,当管内总体积大于10μl时,同比例扩大反应体系。
|
试剂 |
使用量 |
|
反应液 |
10μl |
|
5×PrimeScript II Buffer |
4μl |
|
RNase Inhibitor |
0.5μl |
|
PrimeScript II RTase |
1μl |
|
ddH2O |
Up to 20μl |
3.2.4缓慢混匀后,将含有上述反应液的离心管放入金属加热器中,45℃反应60min,再75℃反应15min,冰上冷却,即为总RNA反转录后的cDNA,-20℃保存。
3.3 PCR扩增
3.3.1第一轮PCR
6.3.1.1以cDNA为模板进行第一轮PCR反应,为了确定最佳模板使用量,分别使用0.5μl,1μl,2μl,3μl,4μl,5μl的oligo dT Primer和random 6-mer的cDNA作为模板,按照下表配置PCR反应体系:
|
试剂 |
使用量 |
|
cDNA |
0.5-5μl |
|
AlpVh-L/CALL 002 |
2μl/2μl |
|
dNTP Mix |
4μl |
|
10×ExTaq Buffer |
5μl |
|
HS Ex Taq |
0.25μl |
|
ddH2O |
Up to 50μl |
按照下表配置并进行PCR反应:
|
循环 |
温度 |
时间 |
|
步骤1 |
98℃ |
3min |
|
步骤2 |
94℃ |
50s |
|
55℃ |
30s |
|
|
72℃ |
40s+2s/cycle |
|
|
转到步骤2 |
23 cycles |
|
|
步骤3 |
72℃ |
5min |
|
步骤4 |
4℃ |
forever |
3.3.1.2反应结束后,取20μl PCR产物进行1%琼脂糖凝胶电泳,最终选取电泳结果中目的条带单一且片段大小为600bp的模板量为最佳模板量,将所有cDNA按照此模板量并使用3.3.1.1相同条件进行PCR反应;
3.3.1.3将所有PCR产物进行1%琼脂糖凝胶电泳,并切胶回收目的片段大小在600bp左右的条带;
3.3.1.4使用通用型DNA纯化回收试剂盒对切胶的胶条进行DNA回收,根据试剂盒说明书,按照下述步骤进行:
(1)柱平衡:向放入收集管中的吸附柱CB2中加入500μl平衡液BL,12000 rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中;
(2)将从琼脂糖凝胶中切下的DNA条带放入干净的离心管中,称取重量;
(3)向胶块中加入等体积PC溶液(如果凝胶重为0.1g,其体积可视为100μl,则加入100μlPC溶液),50℃水浴放置10 min左右,其间不断温和地上下翻转离心管,以确保胶块充分溶解;
(4)将上一步所得溶液加入平衡过的放入收集管中的吸附柱CB2中,12000rpm离心1min,倒掉收集管中的废液,将吸附柱CB2放入收集管中;
(5)向吸附柱CB2中加入600μl漂洗液PW(使用前请先检查是否已加入无水乙醇),12000rpm离心1min,倒掉收集管中的废液,将吸附柱CB2放入收集管中;
(6)重复操作步骤(5);
(7)将吸附柱CB2放入收集管中,12000rpm离心2min,尽量除去漂洗液,将吸附柱置于室温放置2min,彻底晾干;
(8)将吸附柱CB2放入一个干净离心管中,向吸附膜中间位置悬空滴加30μl的ddH2O,室温放置2min。12000 rpm离心2 min,收集DNA溶液;
(9) 将所有纯化回收产物收集至一个离心管中,即为第一轮PCR扩增产物,-20℃保存。
3.3.2第二轮PCR
3.3.2.1将第一轮PCR扩增产物作为模板进行第二轮PCR反应,为了确定最佳模板使用量,分别使用0.1μl,0.2μl,0.3μl,0.5μl,1.0μl,2.0μl作为模板,按照下表配置PCR反应体系:
|
试剂 |
使用量 |
|
第一轮PCR回收产物 |
0.1-2μl |
|
Rvhh FP/RvhhRP |
2μl/2μl |
|
dNTP Mix |
4μl |
|
10×ExTaq Buffer |
5μl |
|
HS Ex Taq |
0.25μl |
|
ddH2O |
Up to 50μl |
按照下表配置并进行PCR反应:
|
循环 |
温度 |
时间 |
|
步骤1 |
98℃ |
3min |
|
步骤2 |
94℃ |
50s |
|
55℃ |
30s |
|
|
72℃ |
40s |
|
|
转到步骤2 |
11cycles |
|
|
步骤3 |
72℃ |
5min |
|
步骤4 |
4℃ |
forever |
3.3.2.2反应结束后,取20μl PCR产物进行1%琼脂糖凝胶电泳,最终选取电泳结果中目的条带单一且片段大小为600bp的模板量为最佳模板量,将已获得的第一轮PCR扩增产物的1/20体积共576个反应按照此模板量并使用3.3.2.1相同条件进行PCR反应;
3.3.2.3使用通用型DNA纯化回收试剂盒对PCR反应液进行DNA纯化,根据试剂盒说明书,按照下述步骤进行:
(1)柱平衡:向放入收集管中的吸附柱CB2中加入500μl的平衡液BL,12000rpm离心1min,倒掉收集管中的废液,将吸附柱重新放回收集管中;
(2)根据PCR反应液的体积,向其中加入等倍体积PC溶液,充分混匀;
(3)将上一步所得溶液加入一个放入收集管中的吸附柱CB2中,室温放置2min,12000rpm离心1min,倒掉收集管中的废液,将吸附柱放入收集管中;
(4)向吸附柱CB2中加入600μl漂洗液PW(使用前请先确认已加入无水乙醇),12000rpm离心1分min,倒掉收集管中的废液,将吸附柱CB2放入收集管中;
(5)重复操作步骤(4);
(6)将吸附柱CB2放入收集管中,12000rpm离心2min,尽量除去漂洗液,将吸附柱置于室温放置2min,彻底晾干;
(7)将吸附柱CB2放入一个干净离心管中,向吸附膜中间位置悬空滴加30μl的ddH2O,室温放置2min,12000 rpm离心2 min,收集DNA溶液。
(8)将回收回的所有产到物收集至一个离心管中,即为第二轮PCR扩增产物,同时取2μl用核酸浓度测量仪检测回收产物的浓度并记录,其余产物-20℃保存。
3.4载体与PCR产物酶切连接
3.4.1酶切体系分别如下表。
载体酶切体系:
|
试剂 |
用量 |
|
载体 |
10μg |
|
Bgl 1 |
10μl |
|
3.1 10×Buffer |
50μl |
|
ddH2O |
Up to 500μl |
第二轮PCR扩增产物酶切体系:
|
试剂 |
用量 |
|
第二轮PCR扩增产物 |
4μg |
|
Bgl 1 |
4μl |
|
3.1 10×Buffer |
20μl |
|
ddH2O |
Up to 200μl |
3.4.2 37℃孵育2h,分别补加10μl和2μl限制性内切酶Bgl 1至载体和PCR扩增产物反应中,继续酶切2h;
3.4.3酶切结束后,使用通用型DNA纯化回收试剂盒纯化载体和酶切产物,纯化步骤同3.3.2.3;
3.4.4酶切反应纯化后,分别取2μl用核酸浓度测量仪检测回收产物的浓度,然后按照下表配制连接反应体系:
|
试剂 |
用量 |
|
PCR产物 |
1.6μg |
|
载体 |
4μg |
|
T4连接酶 |
30μl |
|
10×T4 reaction Buffer |
200μl |
|
ddH2O |
Up to 2000μl |
3.4.5将上述连接反应4℃过夜(约16h)孵育;
3.4.6酶切结束后,使用通用型DNA纯化回收试剂盒纯化连接反应液,纯化步骤同3.3.2.3,取2μl用核酸浓度测量仪检测回收产物的浓度并记录,4℃保存。
3.5细菌文库构建
将连接产物进行电击转化后构建含有纳米抗体片段的大肠杆菌文库。
3.5.1取100ng回收连接产物,按照以下步骤进行电击转化:
3.5.1.1从-80℃超低温保存箱取出1管SS320大肠杆菌感受态细胞,冰上放置5min使其融化;
3.5.1.2使用提前预冷的枪头加入100ng连接产物,轻轻吹匀,转移到已经预冷的1mm电转杯中,轻敲电转杯,让混合物流至电转杯底部;
3.5.1.3设定1800V,1mm间距,进行电转,完成后立即加入950μl 37℃预温的SOC培养基,将菌液从电转杯中快速取出至1.5ml离心管中,37℃,200rpm震荡复苏1h;
3.5.2复苏后的菌液按使用2×YT液体培养基进行10倍梯度稀释,共稀释6个梯度,即将复苏后的菌液取100μl稀释到1000μl,再从中取100μl稀释到1000μl,依次类推,共稀释10,100,1000,10000,100000,1000000倍;
3.5.3从10倍梯度稀释中每管取100μl均匀涂布到2×YT固体培养基(Amp),37℃静置培养过夜;
3.5.4数出梯度稀释平板上的菌落数目,并使用公式计算连接效率:
3.5.5同时随机挑选平板上的48个单克隆菌,接种于含有氨苄抗性的1ml 2×YT液体培养基(Amp)的离心管中,37℃培养3h,按照下表配制PCR反应并进行单菌落PCR:
PCR反应体系:
|
试剂 |
使用量 |
|
菌液 |
2μl |
|
FP/RP |
2μl/2μl |
|
dNTP Mix |
4μl |
|
10×Taq Buffer |
5μl |
|
Taq酶 |
0.25μl |
|
ddH2O |
Up to 50μl |
PCR反应条件:
|
循环 |
温度 |
时间 |
|
步骤1 |
98℃ |
3min |
|
步骤2 |
94℃ |
30s |
|
58℃ |
30s |
|
|
72℃ |
30s |
|
|
转到步骤2 |
25cycles |
|
|
步骤3 |
72℃ |
3min |
|
4℃ |
forever |
3.5.6将单菌落PCR产物进行1%琼脂糖凝胶电泳,目的条带片段大小为600bp的菌落认定为阳性克隆,按公式计算连接产物阳性克隆率:
3.5.7按照3.5.1相同方法进行20个电击转化反应,将复苏后的全部菌液集中至一个15ml摇菌管中混匀,取100μl复苏菌液按照3.5.2-3.5.6方法计算连接效率和阳性克隆率,其余复苏菌液则均匀涂布到5个含有100μg/ml Amp和2%葡萄糖的2%琼脂糖的245mm方形培养基平板中,37℃正置过夜培养;
3.5.8取过夜培养的方形板,在每个培养板表面加6ml 2×YT液体培养基,用涂布棒轻轻将5个方形板菌落刮下并将菌液收集至同一个50ml离心管中,加入终浓度为20%的甘油,即为细菌文库。阳性克隆率为按照3.5.6方法计算出的阳性克隆率,库容量按照公式计算;
3.5.9取20μl上述细菌液到980μl 2×YT液体培养基中并使用分光光度计测量OD600,根据以下公式计算记录细菌文库总OD600;将细菌文库菌液进行分装10管,1ml/管,其余保留在50ml离心管中,-80℃保存;
3.5.10从菌落PCR检测的阳性克隆中随机挑选48个克隆送测序,将测序结果中纳米抗体片段翻译成氨基酸序列后进行序列比对,检测细菌文库中序列多样性。
3.6噬箘体文库构建
3.6.1噬菌体扩增
3.6.1.1从-80℃冰箱中取出细菌文库,冰上融化,根据下面公式计算出相应菌液量,转接到100ml含10μg/ml Tet和100μg/ml Amp的2×YT液体培养基中,以使初始OD600为0.1:
3.6.1.2在恒温摇床中37℃,250rpm培养直至菌液OD600达到0.5-0.55;
3.6.1.3加入辅助噬菌体M13KO7以使细菌个数:噬菌体数=1:20:
3.6.1.4在恒温摇床中37℃,220rpm继续培养30 min;
3.6.1.5分别加入终浓度为50μg/mlKana和终浓度为0.2μM IPTG,恒温摇床中30℃,250rpm过夜培养。
3.6.2噬菌体文库制备
3.6.2.1将过夜培养的菌液转移至新的50ml离心管中4000 rpm,4 ℃离心10min;
3.6.2.2将离心后的上清液转移至新的50ml离心管中,加入1/4体积的4℃预冷的20%PEG/2.5M NaCl,充分混匀后冰上放置30min;
3.6.2.3 4000 rpm,4 ℃离心20 min,弃上清,并在纸上倒置2min;
3.6.2.4加入1ml PBS重悬沉淀,将重悬液移至新的1.5ml离心管,12000rpm,4 ℃离心20min;
3.6.2.5将离心后的上清转移至新的1.5ml离心管,加入1/4体积预冷的20%PEG/2.5M NaCl溶液,混匀后冰上放置10min;
3.6.2.6 12000rpm,4 ℃离心10min,弃上清,加入1ml PBS重悬沉淀;
3.6.2.7 12000rpm,4 ℃离心2min,将上清转移到新的1.5ml离心管中,即为噬菌体文库,100μl /管分装,长期-80℃保存,短期(1-2周)可于4℃放置保存。
3.6.3噬菌体文库效价检测
3.6.3.1将-80℃冰箱保存的SS320菌株在2×YT固体培养基(Tet抗性)进行单菌落划线,37℃过夜培养(4℃保存一周),从单菌落板上挑取一个单菌落到5ml含有10μg/ml Tet的2×YT培养基,37℃过夜培养;
3.6.3.2取过夜培养菌液250μl转接到含5ml含有10μg/ml Tet的2×YT液体培养基中,37℃,250rpm培养约45min-60min后至OD600为0.5-0.55;
3.6.3.3取3.7.2方法制备好的噬菌体文库10μl,在1.5ml离心管中10倍梯度稀释,共稀释12个梯度,即将噬菌体文库取10μl稀释至100μl,再从中取10μl稀释至100μl,依次类推,共稀释12个梯度至10-12,振荡混匀;
3.6.3.4在每个稀释离心管中加入90μl的SS320菌液,混匀后37℃孵育30min;
3.6.3.5从每个稀释离心管中取5μl滴加到2×YT固体培养基(Amp)中,37℃过夜培养;
3.6.3.6统计板上可以明显区分单菌落的稀释度的单菌落数量,并按照下面公式计算每毫升噬菌体溶液中噬菌粒的数量,即噬菌体文库效价。
4. 实验结果
4.1总RNA提取和反转录
4.1.1总RNA琼脂凝胶电泳结果:
图1羊驼PBMC总RNA琼脂糖凝胶电泳结果
4.1.2反转录cDNA结果:
4.2 PCR扩增
4.2.1第一轮PCR扩增
4.2.1.1确定最佳扩增模板量
琼脂糖凝胶电泳结果显示,oligo dT Primer转录后进行第一轮扩增使用2μl作为模板扩增条带强清晰且无杂带,选择2μl为最佳扩增模板量;random 6-mers转录后进行第一轮扩增使用2μl作为模板扩增条带清晰且无杂带,选择2μl为最佳扩增模板量。

图2使用不同cDNA模板量的琼脂糖凝胶电泳
4.2.1.2第一轮PCR扩增和回收
|
引物 |
项目 |
值 |
|
Oligo dT Primer |
单个反应模板量 |
2μl |
|
反应个数 |
130 |
|
|
Random 6-mers |
单个反应模板量 |
2μl |
|
反应个数 |
130 |
|
|
合并后 |
回收后DNA体积 |
1080μl |
|
回收后DNA浓度 |
55.5 ng/μl |
|
|
回收后DNA总量 |
59.9ug |
4.2.2第二轮PCR扩增
4.2.2.1确定最佳扩增模板量
琼脂糖凝胶电泳结果显示,第二轮扩增使用0.1μl作为模板扩增条带清晰且无杂带,选择0.1μl为最佳扩增模板量。

图3使用不同的第一轮PCR产物模板量的琼脂糖凝胶电泳
4.2.2.2第二轮PCR扩增和回收
|
项目 |
值 |
|
|
每个反应模板量 |
0.1μl |
|
|
反应个数 |
576 |
|
|
合并后 |
回收后DNA体积 |
2760μl |
|
回收后DNA浓度 |
241.4ng/μl |
|
|
回收后DNA总量 |
666.3μg |
4.3载体和PCR产物酶切、连接
4.3.1酶切、连接检测结果
|
项目 |
值 |
|
|
载体酶切回收 |
使用载体量 |
10μg |
|
回收后体积 |
50μl |
|
|
回收后浓度 |
80.7ng/μl |
|
|
回收后总量 |
4.0μg |
|
|
PCR产物酶切回收 |
使用PCR产物量 |
4μg |
|
回收后体积 |
50μl |
|
|
回收后浓度 |
55.5ng/μl |
|
|
回收后总量 |
2.8μg |
|
|
连接产物回收 |
回收后DNA体积 |
50μl |
|
回收后DNA浓度 |
114.1ng/μl |
|
|
回收后DNA总量 |
5.7ug |
4.4细菌文库构建
4.4.1细菌文库检测
|
项目 |
值 |
|
连接效率 |
1.2×108pfu/100ng |
|
库容量 |
2.3×109pfu |
|
阳性克隆率 |
96% |
|
细菌库体积 |
50 ml |
|
细菌库OD600 |
77.88 |
4.4.2阳性克隆率菌落PCR
菌落PCR琼脂糖凝胶电泳结果显示,挑选的55个单克隆有53个为阳性克隆,阳性率为96%,细菌文库阳性克隆率符合要求。
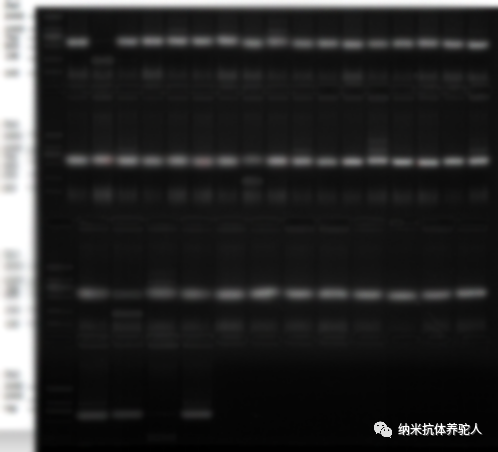
图4菌落PCR琼脂糖凝胶电泳结果
4.4.3序列多样性
随机挑选48个单克隆测序后使用GENtle软件转录翻译成蛋白质序列,序列多样性比对显示48个序列均为独立序列,多样性100%,细菌文库多样性符合要求要求。

图5序列多样性比对
4.5噬箘体文库制备
4.5.1噬菌体文库检测
|
项目 |
值 |
|
体积 |
1.0 ml |
|
效价 |
4.8×1012pfu/ml |
咨询
- 908
- 点赞
- 复制链接
- 举报