
空间转录组测序
- shbio
- 伯豪生物
- 上海市
- 现货
- 按需定制
- 议价
- 2025-01-06 15:20:32
上海伯豪生物技术有限公司
- 英文名称
- Spatial transcriptome sequencing
- CAS号
- -
- 别名
- 空间转录组测序/空间基因表达测序
- 关键词
- 空间转录组测序空间基因表达空间基因测序空间转录组测序服务空间转录组测序价格
- 科研服务
- 空间转录组测序/空间基因表达测序

基因表达具有时间和空间的特异性,通过对不同时间点的样本取材,使用单细胞转录组测序技术能够解析时间维度上细胞类型和基因表达的变化过程。然而单细胞测序实验的前提是组织必须通过机械分离或酶解消化成单细胞悬液,此过程不可避免的丢失了组织中细胞所处的原始位置信息,也导致了细胞间的通讯网络被打破,这使我们难以获得组织中不同区域的细胞构成和基因表达状态,以及不同功能区之间的基因差异表达等信息。现有的原位表达图谱主要是通过报告基因或原位杂交等技术来实现,但是这些方法实现比较困难,并且通量低,限制了多样本、高时效的应用。而空间转录组技术则可以高 效的检测组织中空间原始位置上的基因表达模式。
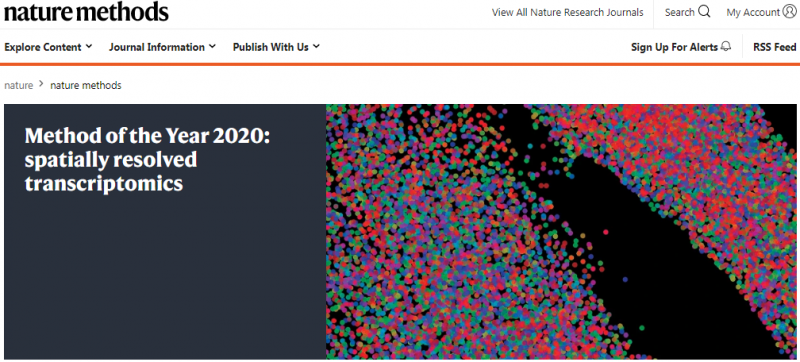
▲图 空间转录组被评为 2020 年度技术
注:图片来源互联网 - 侵删
空间转录组(Spatial Transcriptomics)就是将基因的表达情况与关注的组织切片的免疫化学染色图像进行整合,从而将组织内不同细胞的基因表达信息定位到组织的原始空间位置上去,进而直接观测组织中不同部位功能区基因表达的差异。空间转录组技术利用了常规的原位技术和组学技术两方面的优势。实际上空间转录组已不是新名词,2016 年 Joakim Lundeberg 的 Spatial Transcriptomics 技术在 Science 上发表,2017 年景乃禾老师的 GEO-seq 技术在 Nature Protocols 上发表 [2]。目前已发表的关于空间转录组技术有主要有 Spatial Transcriptomics, Slide-seq, LCM-seq, seqFISH, MERFISH, Liver single cell zonation, Geo-seq 和 Tomo-seq, 涉及物种有人、小鼠、果蝇、秀丽隐杆线虫、斑马鱼、拟南芥、杨树和云杉等。
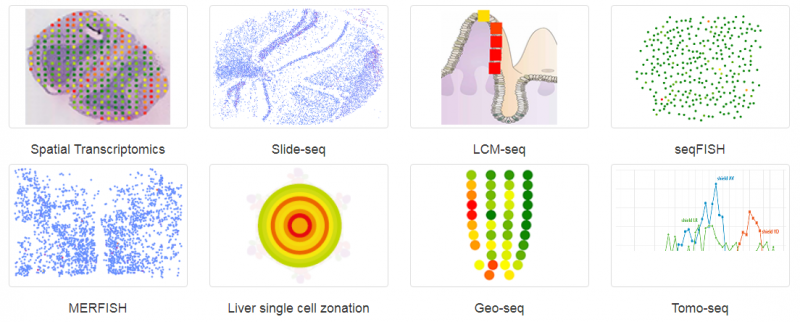
▲图 常见空间转录组技术
注:图片来源互联网 - 侵删
其中,瑞典皇家理工学院的 Joakim Lundeberg,基于芯片和空间条形码技术发明了高通量的空间转录组测序方法,并创建了 Spatial Transcriptomics 公司。2018 年底 10X Genomics 宣布收购 Spatial Transcriptomics,并于 2019 年发布 Visium 空间基因表达解决方案(Visium Spatial Gene Expression Solution)。10X Genomics 公司提供的 Visium 空间基因表达解决方案是高通量空间转录组的商业化解决方案,其可以检测完整组织切片的总 mRNA,将总 mRNA 的空间信息与形态学内容相结合,并绘制所有基因表达发生的位置,获得完整的基因表达图谱;在确定不同细胞群的同时保留空间位置,为细胞功能、表型和组织微环境中位置的关系提供了重要信息。

空间转录组学技术优势:是转录组学研究领域的新方向,也是研究细胞异质性方面的新方法。
(1)准确定位:探针有效定位组织中 RNA 空间位置,实时了解组织中的转录组天然状态。
(2)简单易行:芯片设计简单易操作、重复性高、可快速获得高分辨率空间转录组信息。
(3)适用性广:有效应用于发育生物学、肿瘤生物学、脑神经科学、植物研究等各生物学领域。
1、伯豪个性化方案:空间转录组测序研究需求千差万别,伯豪生物专业科研团队针对客户需求一对一项目建议,为客户定制空间基因表达解决方案。
2、组织保存液:空间转录组技术对样本质量要求高,新鲜组织样本需要立即冷冻包埋才能更好的保持样本 RNA 质量,从而保障实验准确性。伯豪生物自研了伯优®组织保存液,4℃条件下新鲜组织样本离体 48 小时,细胞活性及细胞形态结果无明显影响,有效解决样本采集→保存运输→冷冻包埋的不良影响。
3、高质量组织切片制备:伯豪生物科研服务团队经大量项目样本经验累积,探索出 对多种类型组织制备出高质量切片方法:“不同组织类型的切片需要优化不同的条件”。
4、全面的生信分析流程和个性化数据分析。

将冷冻组织切片放置在空间转录组芯片的捕获区域内,进行 HE 染色和成像后,对组织切片进行透化处理,细胞内的 mRNA 释放,从而被芯片上带有 oligo-dT 的探针捕获,并且每个探针都带有特异的位置信息(Spatial barcode),然后以 mRNA 为模版进行 cDNA 合成,构建文库后再通过测序,获得基因表达信息的同时,每一条测序 reads 因带有 Spatial barcode,从而能够获得基因表达的位置信息。
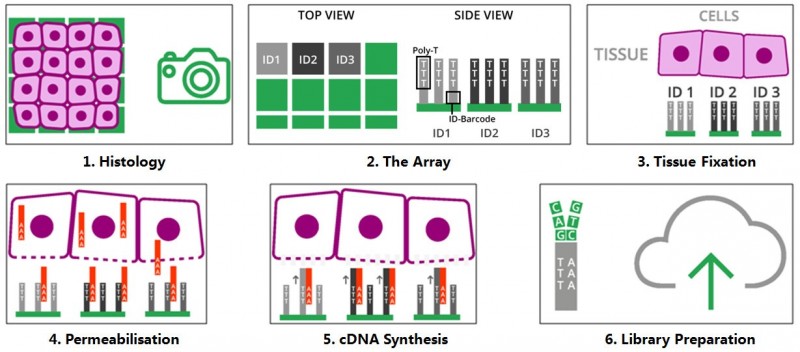
▲图 空间转录组技术的原理
注:图片来源互联网 - 侵删
空间转录组技术包含两种芯片,分别为组织优化芯片(Tissue Optimization)和基因表达芯片(Library Preparation)。组织优化芯片用来摸索组织透化的时间,基因表达芯片用来进行正式样本的空间转录组实验。其中基因表达芯片上有 4 个捕获区域,每个区域大小为 6.5mm×6.5mm,每个捕获区域中有 5000 个带有特异地址序列的探针簇,称为 barcoded spots,每个 spot 直径为 55um,包含数百万个用于捕获的 oligo 探针序列,相邻两个 spot 的中心距离为 100um。探针序列的结构为:测序引物结合序列,16nt 的位置序列,12nt 的 UMI 序列以及 30nt 的 oligo-dT 序列。

▲图 空间转录组两种芯片
注:图片来源互联网 - 侵删
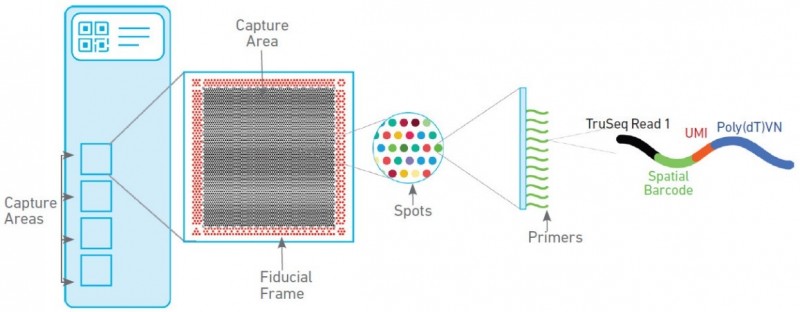
▲图 空间转录组基因表达芯片工作原理
注:图片来源互联网 - 侵删

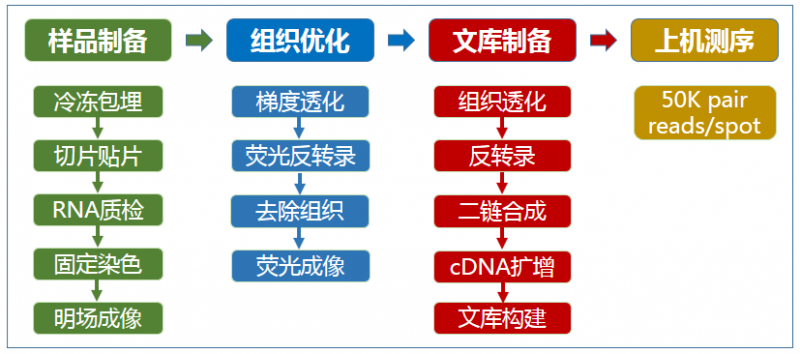
▲图 空间转录组测序实验流程

方法一:组织冷冻(不常用)
1、异戊烷和液氮浴:如图所示,不锈钢烧杯内倒入异戊烷至 2 / 3 体积,然后将不锈钢烧杯放入液氮中(与异戊烷液位相同),孵育 15 分钟。
2、新鲜样本可用 PBS 冲洗,去除残留血液,然后使用实验室纸巾吸干组织表面多余的血液或液体,防止冰晶的形成(注:组织长和宽不可超过 6mm±0.2mm,否则后续实验无法贴片)。
3、用镊子或刮刀将组织整体浸没在异戊烷中,直至整体冰冻(注:冷冻时间可根据组织类型和大小而改变)。
4、冷冻后,取出组织转移到预冷的密封容器中,干冰转移至 -80°长期保存或立即进行下一步(注:为防止组织样品蒸发和脱水,冷冻的组织样品必须储存在密封容器中以长期保存)。
方法二:冷冻组织包埋(常用)
1、粉状干冰:用研钵和杵准备干冰粉。
2、冷冻 OCT:将 OCT 放在冰上≥30 分钟(注:OCT 请使用指定品牌)。
3、预冷镊子:将镊子放在干冰中≥30 分钟。
4、包埋盒上需标记组织样本的方向(注:在添加 OCT 和组织之前,需先在包埋盒上标记,因为一旦冻结,OCT 将迅速变成白色,这使得以后很难确定组织方向)。
5、用冷却的 OCT 铺平包埋盒底部,避免产生气泡。
6、从不锈钢烧杯中取出冰冻的组织,加入到 OCT 的包埋盒中心位置,继续倒入 OCT 覆盖样本。需避免气泡产生,尤其在组织附近。
7、立即将含有组织和 OCT 的包埋盒放在干冰粉上,直至整体冻结(约 20min 以上)。
8、将包埋盒放置到密封袋中,干冰运输。
注意事项
1、新鲜组织样本包埋
目前新鲜组织的包埋方法有两种,一种是液氮 + 异戊烷法;另一种的干冰法。对于临床手术切下来的组织样本一般使用干冰的包埋方法。对于穿刺样本等一些较小,较轻的样本,一般推荐用液氮 + 异戊烷的方法进行冷冻。OCT 包埋组织块可以在–80ºC 的密封容器中长期保存,或立即进行冷冻切片。
2、冷冻切片切、组织样本质控及贴片
由于空间转录组检测的是组织中的 RNA,因此要对切片中的 RNA 质量进行检测。我们一般取 10 片组织切片进行 RNA 抽提并质检,确定组织中 RNA 完整性(RIN>7)。所以要求我们的组织样本要保证可以切到至少 20 片 10um 厚度的切片,以便完成所有的实验。
3、组织优化
组织优化的目的是摸索样本的透化条件,保证组织切片中的 mRNA 能够充分释放。该步骤是获取真实实验结果的必要条件。否则,我们无法判断是基因表达高低到底是因为透化不充分导致的,还是实际就是这个样子。因此:每个样本建议都要做透化,尤其是临床样本。
4、成像
应使用 Visium Imaging Test Slide 验证成像设置。明场成像基准框和基准标记应清晰可见,并使用 Brightfeld 设置聚焦。Visium Imaging Test Slide 四个区域(A1,B2,C1,D2)具有荧光斑点,可通过 TRITC 和 Cy5 flter cubes 检测到,荧光设置应清晰可见 A1,B2,C1 和 D2 中的荧光点,且荧光点信号应从左到右减小。
5、正式实验
正式实验时要对反转录后的 cDNA 长度分布,浓度和量进行判断。cDNA 的长度分布在~200bp-9000bp 之间,在 1000bp 左右会有峰值(不同的组织类型会有些许差异)。
6、测序
在捕获区域,每个组织覆盖的 spot 建议至少测 50000 read pairs。整个捕获区域共有 5000 个 spots。可以根据组织贴到芯片上后,覆盖芯片的大小来判断测序的数据量。
计算公式(Coverage Area x total spots on the Capture Area)x 50,000 read pairs/spot 例如:组织覆盖了 60% 的区域,则数据量为(0.60 x 5,000 total spots) x 50,000 read pairs/spot=150 million read pairs。

图 切片覆盖芯片区域百分比

空间转录组数据分析的核心是根据每个芯片上每个 spot 的基因表达信息进行聚类,然后将 spot 根据坐标位置序列放回到组织的图像上,同时可以对每个 gene 在组织上表达的空间位置进行定位。
获得测序数据后,首先利用 Space Ranger 软件可以自动化的对图像进行处理、数据比对和 Barcode 处理。另外一个软件 Loupe Cell Browser 是一个适用于 Windows 和 MacOS 的桌面应用程序,它可以快速、轻松地可视化和分析 10X Visium 数据。伯豪生物除了提供 spot 基因数和 UMI 数统计、切片 spot 聚类和聚类亚群 marker 基因分析等基础和高级分外,同时还提供个性化分析,如特定 pathway 功能富集分析等。
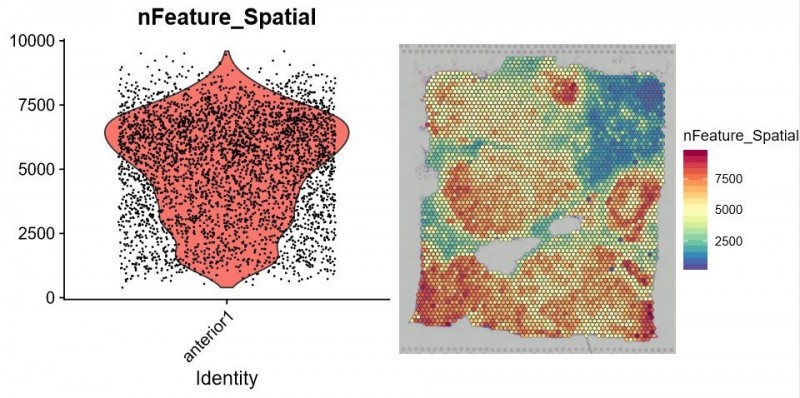
▲图 每个 spot 特异表达的基因数统计
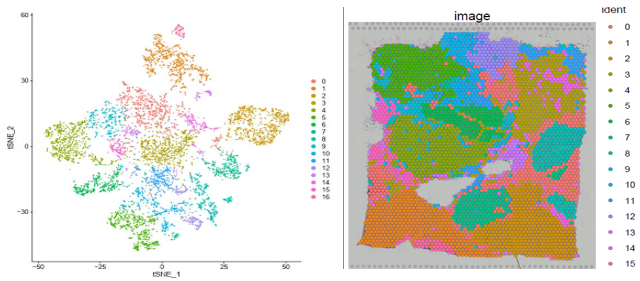
▲图 聚类结果及切片 spot 位置分布展示
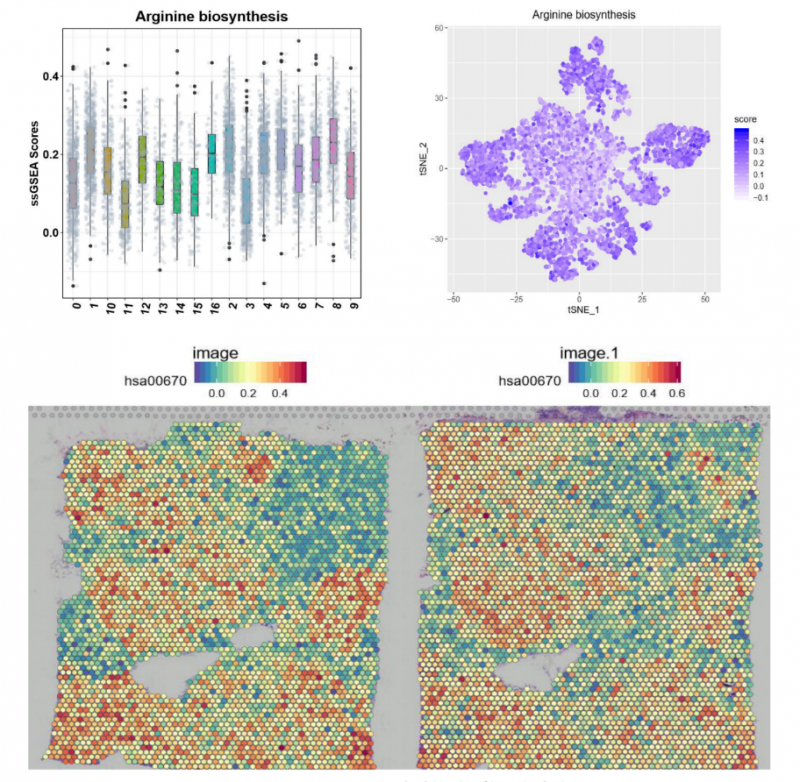
▲图 特定 pathway 功能富集分析
结合组织区域分布对数据进行挖掘
大部分组织其实是有其特定的区域划分的,比如说大脑里有皮层、丘脑、海马、脉络丛等多个区域。将组织的区域划分和亚群(或细胞类型)的分布结合起来还是能发现很多有价值的信息的。
可以根据不同区域特异表达的 maker 基因的分布来判断每个区域在组织切片上的位置。例如皮层 marker 基因 STX1A 的表达分布,海马 marker 基因 HPCA 的表达分布等。
结合病理学特征对数据进行挖掘
空间转录组技术正真的精髓不是研究细胞亚群的分布,而在于将它在空间位置上体现的异质性跟组织病理学特征的分布进行结合,挖掘在不同病理学特征下转录组学的差异。这对于研究疾病病变的机制、帮助临床实现更好的患者分子分型、以及空间位置 Biomarker 的挖掘方面都是非常有价值的。通过手动把这些区域圈出来进行转录组层面的比较,找出不同病灶区的特异性 marker,分析疾病在一步步发展进程中生物学功能的变化,甚至可以思考一下是否能找出一些关键性因子来阻断疾病的进展。
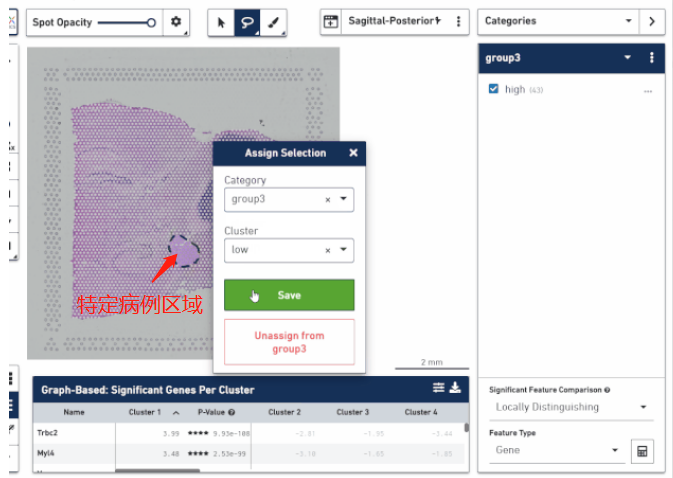
▲图 根据病理信息选取特定区域分析
空间转录组联合单细胞 RNA 测序解析细胞类型的空间位置信息(Multimodal intersection analysis,MIA)
空间转录组测序可以获得不同基因在组织切片上的空间位置信息,但不能获得详细的细胞类群信息(空间转录组不是单细胞分辨率,只能粗略的分析切片上不同位置的细胞类型)。因此,需要借助但细胞测序数据来分析细胞类型,然后通过生物信息学的分析方法将单细胞类群映射到空间转录组数据上。
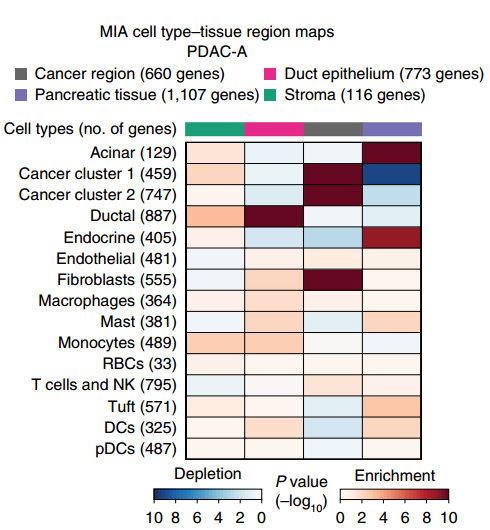
▲图 MIA 热图
备注:MIA 热图,上方的颜色条反映了 ST 区域的子聚类(cancer region,Pancreatic tissue,Duct epithelium 和 stroma)。左侧代表不同的细胞类群。色块代表 enrichment 或者 depletion。Enrichment 代表该细胞类群富集到了该区域。Depletion 代表该细胞类群在该区域缺失。

空间位置信息,或者细胞在组织中天然的状态在研究过程中其实具有十分重要的价值,特别针对某些研究领域,如发育生物学(不同位置的细胞接受不同的信号浓度梯度、响应不同的外界刺激,具有不同的发育命运)、肿瘤生物学(肿瘤组织与癌旁组织的区别,肿瘤细胞侵润过程中肿瘤细胞的变化与对正常细胞的影响,肿瘤转移的不同过程阶段等)、脑神经科学(不同脑区位置的神经元结构、神经连结,中间神经元投射,突触前后,神经胶质相互影响等等),细胞来源的位置信息是极为关键的决定因素。
空间转录组测序技术除了在人和动物上得到了广泛的应用之外,在植物领域也有所突破。2017 年发表在 nature plant 上的一篇文章就阐释了空间转录组在拟南芥中的应用,利用空间数据作者分析了 141 个基因的表达水平差异,8 个花序组织域中 189 条通路。伯豪生物作为较早的空间转录组测序技术服务商,也在植物的空间转库组领域有所突破。开发了多种植物组织的空间转录组样本制备,透化条件摸索等。获得了宝贵的项目经验。
常见空间转录组的应用方向主要在肿瘤学,免疫学,发育生物学,神经科学及病理学等方向。
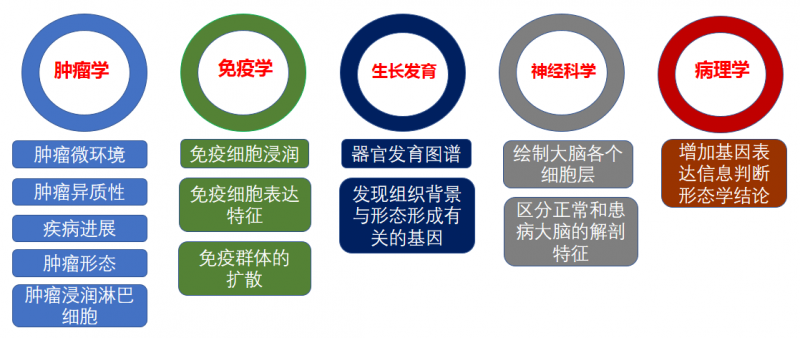
▲图 空间转录组的应用方向
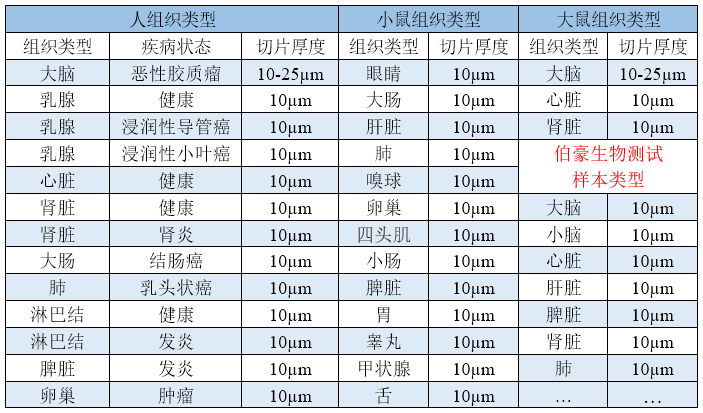
▲表 已做过优化的样本类型

案例一
空间转录组学联合单细胞 RNA-seq 揭示胰腺导管腺癌的组织结构
Integrating microarray-based spatial transcriptomics and single-cell RNA-seq
reveals tissue architecture in pancreatic ductal adenocarcinomas [5]
发表杂志:Nature biotechnology
影响因子:36.558
发表时间:2020 年 1 月
单细胞 RNA 测序(single RNA sequence,scRNA-seq) 能够系统地识别组织中的细胞类群,但它不能获取各个细胞类型在组织中的空间位置信息。空间转录组测序(spatial transcriptomics,ST),可以获得不同基因在组织切片上的空间位置信息,但不能获得详细的细胞类群信息。作者通过对胰腺导管癌病人的同一样本同时进行 scRNA 测序和 ST 测序。同时开发了一种多模态交叉分析方法(Multimodal intersection analysis,MIA)来将单细胞测序数据映射到空间转录组数据上,获得各种细胞类型在组织上的空间分布。研究发现导管细胞、巨噬细胞、树突状细胞和癌细胞的亚群在不同的空间位置区域上富集或丢失,以及与其他类型的细胞有显著的共富集。此外,作者还发现了炎症成纤维细胞和表达应激反应基因模块的癌细胞的定位。
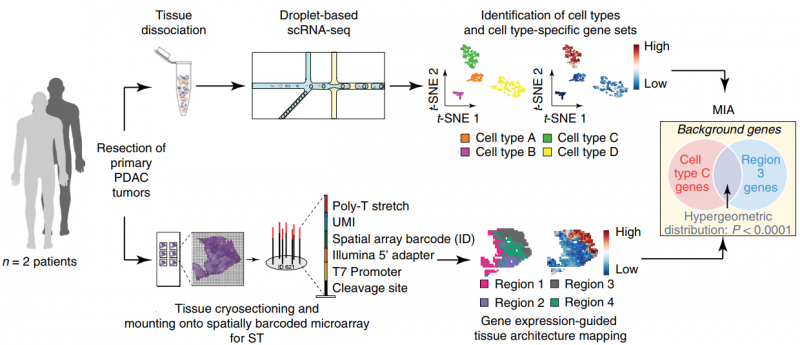
图:ST 联合 scRNA 揭示胰腺导管癌组织细胞类型及结构
案例二
空间转录组联合单细胞测序揭示心脏发育
A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart[11]
发表杂志:Cell
影响因子:38.637
发表时间:2019 年 12 月
摘要:人类心脏形态发生的过程尚不清楚。它的完整特性需要用单细胞空间分辨率深入探索器官范围内基因表达的协调。在这里,我们提出了一种分子方法,揭示了在三个发育阶段的胚胎心脏细胞类型的转录景观,并将细胞类型特异性基因表达映射到特定的解剖结构域。空间转录组学鉴定了在每个发育阶段对应不同解剖区域的独特基因谱。作者通过对人类心脏发育有三个阶段:孕早期 4.5- 5 周,6.5 周和怀孕后 9 周的人类胚胎心脏样品进行单细胞 RNA 测序鉴定的人类胚胎心脏细胞类型,同时通过空间转录组测序技术来获取基因表达的空间位置信息。然后使用原位测序来细化这些结果,并为三个发育阶段创建一个空间亚细胞图谱。形成了一个公开的人类心脏发育的网络资源,以促进未来对人类心脏发生的研究。
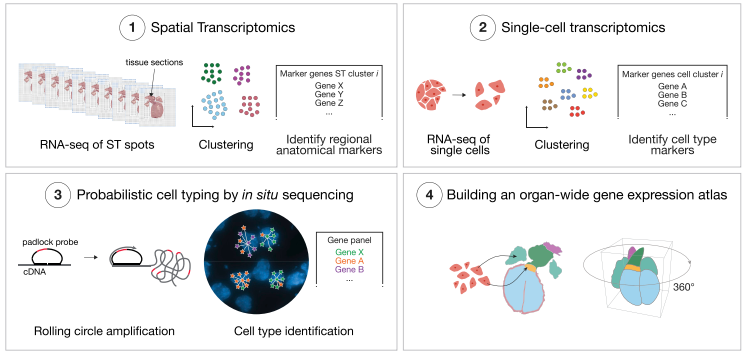
图 18 实验设计思路
A、本研究包括三种人类心脏组织的发育阶段。B、本研究所采用的分子生物学方法:(1)利用空间转录组技术(ST),鉴定出了解剖学区域特异的 marker;(2)用 scRNA-seq 技术分析心脏中间时间点的细胞类型异质性;(3)用 ISS 技术定位亚细胞分辨率的关键基因;(4)构建器官水平的 3D 基因表达图谱。
案例三
单细胞 RNA 测序 & 空间转录组测序“强强联手”揭示肠道发育
Spatiotemporal analysis of human intestinal development at single-cell resolution
发表期刊:Cell
影响因子:38.637
发表时间:2021 年 1 月
肠道是人体大的屏障器官,与肠道微生物共生协调营养需求和免疫。多种相互关联的细胞类型构成了成熟的肠道及其不同的形态,但其发育的分子基础仍不清楚。来自牛津大学 Simmons 和 Koohy 教授团队通过单细胞测序联合空间转录组技术来研究肠道发育过程中形态的变化。研究对来自 17 例胚胎的 77 个样本进行了单细胞 RNA 测序(使用寡核苷酸标记抗体的多重混样技术)以及对来自 5 个样本的 8 张切片进行了空间转录组测序。通过一系列的生物信息学分析方法,包括细胞间通讯网络构建,SCENIC 转录因子模块分析,拟时序分析,RNA 速率分析,基因功能富集分析等,共鉴定了 101 种细胞类型,包括上皮细胞和间充质祖细胞群和与关键形态发生的重要程序。作者描述了隐窝 - 绒毛轴形成的原理,发育中的肠道的神经、血管、间充质形态发生和免疫群体。确定了发育中的成纤维细胞和肌成纤维细胞亚型的分化层次,并描述了它们的不同功能,包括作为血管生态位细胞的功能。作者确定了 Peyer’s patches 和 gut-associated lymphoid tissue (GALT) 的起源,并描述了位置特异性免疫程序。提出了一个无偏倚的分析形态梯度,可以用来指引连续的细胞分化、定义细胞、以及罕见的发育性肠道疾病相关的位置区域。此外,作者还编制了一个公开的胎儿肠道在时间和空间层面发育(STAR-FINDer) 的在线数据库,以促进进一步的工作。
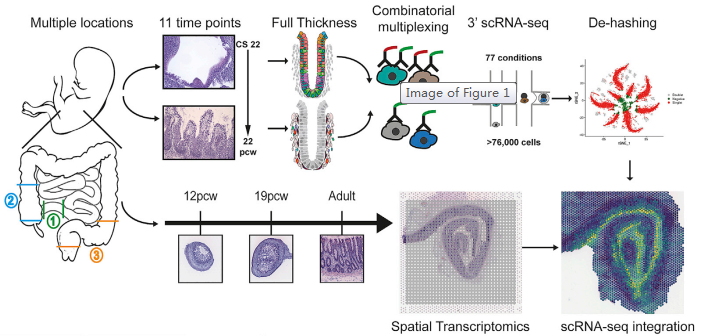
图 研究概况
案例四
空间转录组测序揭示阿尔兹海默疾病机制
Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease
发表期刊:Cell
影响因子:38.637
发表时间:2020 年 8 月
虽然在阿尔茨海默病(AD) 淀粉样斑块周围观察到复杂的炎症样改变,但对这种反应的分子变化和细胞相互作用知之甚少。在 AD 小鼠模型中,作者利用空间转录组学技术研究淀粉样斑块周围直径为 100 毫米的组织结构域发生的转录变化。研究证实了髓磷脂和少突胶质细胞基因(OLIGs) 富集的基因共表达网络的早期改变,而涉及补体系统、氧化应激、溶酶体和炎症的斑块诱导基因(猪)的多细胞基因共表达网络在疾病的后期显著。此外,在小鼠和人类大脑切片上使用原位测序确认了大多数在细胞水平上观察到的改变。空间转录组学分析为解开 AD 和其他脑部疾病致病特征附近的失调细胞网络提供了一种新的方法。
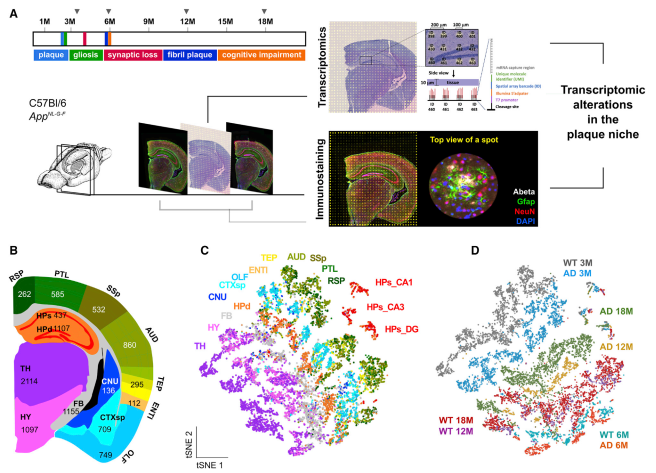
图 成年 AD 小鼠大脑的空间解析转录组谱
案例五
空间转录组图谱为前列腺癌提供异质性的新视图
Spatial Maps of Prostate Cancer Transcriptomes Reveal an Unexplored
Landscape of Heterogeneity
发表期刊:Nature communication
影响因子:12.212
发表时间:2018 年 9 月
前列腺癌包含大量的肿瘤内异质性,在原发肿瘤和远距离转移中都存在基因改变前列腺癌的病理严重程度,尽管有分子标记和核磁共振的进展,但通常是根据 Gleason 分级(Gs)系统来评分的,该系统仅使用组织学数据,经常在血液和肿瘤分期中补充 PSA 测量。然而,这种分类方法有局限性,并提出了新的备选方案。利用一种新的反卷积方法,作者分析了近 6750 个组织区域的转录组,并提取了不同组织成分的不同表达谱,如间质、正常和针腺、免疫细胞和癌症。同时区分了健康区域和病变区域,从而对前列腺癌进展过程中的基因表达变化提供了见解。与病理学家的注释相比,空间转录组可以更准确地描述了癌灶的范围,有趣的是,与组织学变化无关。
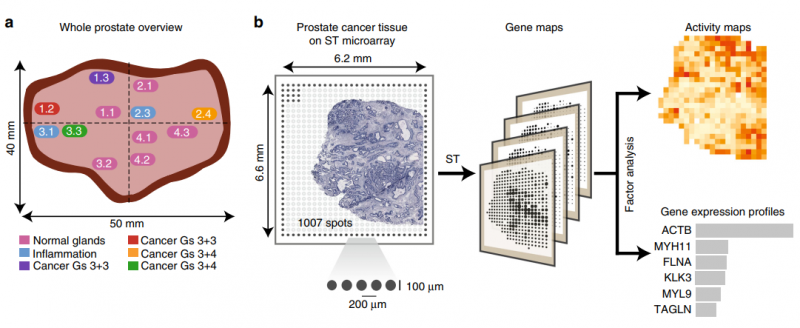
图 前列腺癌的空间转录组学(ST) 研究设计
案例六
空间转录组解析模式植物组织基因表达的空间信息
Spatially resolved transcriptome profiling in model plant species
发表期刊:Nature plant
影响因子:13.256
发表时间:2017 年 5 月
要理解复杂的生物系统,需要对特定组织域进行功能研究。然而,现有的空间转录组测序技术仅适用于有限范围的生物,主要是哺乳动物。在这里,作者提出了可用与植物组织的方法,在空间分辨率的状态下来广泛的解析模式植物系统。该过程包括高通量的空间转录组分析,然后是空间基因和通路分析。作者首先从模型被子植物和裸子植物的显微切片中生成空间转录组谱,证明了该技术的可行性。在拟南芥中,利用空间数据分析了 8 个花序组织结构域中 141 个基因和 189 条通路的表达水平差异。作者通过将空间转录组学技术和功能谱分析技术相结合这一新策略,应用于广泛的植物物种,该技术将是一种解决发育和进化生物学基本问题的关键方法。
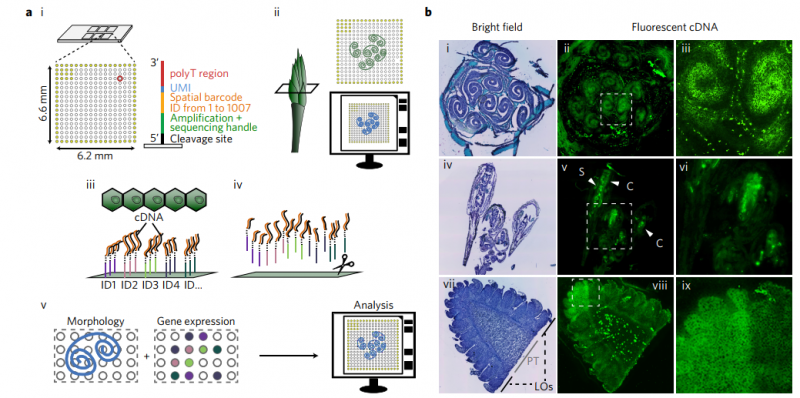
▲图 植物的空间解析转录组分析

[1]. Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353(6294):78-82.
[2]. Peng G, Suo S, Chen J, et al. Spatial Transcriptome for the Molecular Annotation of Lineage Fates and Cell Identity in Mid-gastrula Mouse Embryo. Dev Cell 2016, 36(6):681-697.
[3]. https://www.spatialomics.org/SpatialDB/
[4]. Michaela Asp, Stefania Giacomello, et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart[J]. Cell, 2019, 179, 1647–1660.
[5]. Maniatis S, Äijö T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in
amyotrophic lateral sclerosis[J]. Science, 2019, 364(6435): 89-93.
[6]. Carlberg K, Korotkova M, Larsson L, Catrina AI, Stahl PL, Malmstrom V. Exploring inflammatory signatures in arthritic joint biopsies with Spatial Transcriptomics[J]. Scientific reports. 2019,9(1):18975.
[7]. José, Fernández, Navarro, et al. ST viewer: a tool for analysis and visualization of spatial transcriptomics datasets.[J]. Bioinformatics (Oxford, England), 2019.
[8]. Berglund E , Maaskola J , Schultz N , et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity[J]. Nature Communications, 2018, 9(1).
[9]. ST Spot Detector: a web-based application for automatic spot and tissue detection for spatial Transcriptomics image datasets[J]. Bioinformatics, 2018.
[10]. Anna L , Natalija G , Tove B , et al. Gene expression profiling of periodontitis-affected gingival tissue by spatial transcriptomics[J]. Scientific Reports, 2018, 8(1):9370.
[11]. Kim T , Hanna E , Jonas M , et al. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma[J]. Cancer Research, 2018:canres.0747.2018.
[12]. Salmen F, Stahl PL, Mollbrink A, Navarro JF, Vickovic S, Frisen J, et al. Barcoded solid-phase RNA capture for Spatial Transcriptomics profiling in mammalian tissue sections[J]. Nature protocols. 2018,13(11):2501-34.
[13]. Giacomello S , Fredrik Salmén, Terebieniec B K , et al. Spatially resolved transcriptome profiling in model plant species[J]. Nature Plants, 2017, 3(6):17061.
[14]. Asp M , Salmén, Fredrik, St?Hl P L , et al. Spatial detection of fetal marker genes expressed at low level in adult human heart tissue[J]. Scientific Reports, 2017, 7(1):12941.
[15]. Stahl P L , Salmen F , Vickovic S , et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics[J]. Science, 2016, 353(6294):78-82.
[16]. Vickovic S , St?Hl P L , Salmén, Fredrik, et al. Massive and parallel expression profiling using microarrayed single-cell sequencing[J]. Nature Communications, 2016, 7:13182.
[17]. Anders, Jemt, Fredrik, et al. An automated approach to prepare tissue-derived spatially barcoded RNA-sequencing libraries.[J]. Scientific reports, 2016.
[18] Berglund E , Maaskola J , Schultz N , et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity[J]. Nature Communications, 2018, 9(1).
[19] Chen W T , Lu A , Craessaerts K , et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease[J]. Cell, 2020.
[20] Giacomell S , Fredrik Salmén, Terebieniec B K , et al. Spatially resolved transcriptome profiling in model plant species[J]. Nature Plants, 2017.
[21] David Fawkner-Corbett, et al. Spatiotemporal analysis of human intestinal development at single-cell resolution[J]. Cell. 2021 Feb 4;184(3):810-826.e23. doi: 10.1016/j.cell.2020.12.016. Epub 2021 Jan 5.